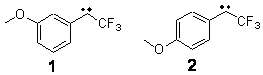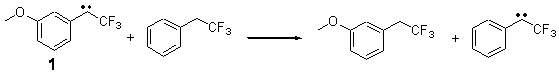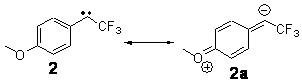D. Janardanan, D. Usharani, S. Shaik Angewandte Chemie International Edition 2012, 51, 4421-4425 (Paywall)
In a Perspective in Nature Chemistry last year1 Sason Shaik and co-workers described bond activation by metal-oxo enzymes and synthetic reagents. In it, they argued that Hund's rule of maximum multiplicity (valid for atoms) has an analogue for reactions and kinetics of (bio)inorganic species: the exchange-enhanced reactivity (EER). Pathways that increase the number of unpaired and spin-identical electrons on a metal center will be favored by exchange interactions, and hence are favored over pathways that keep the same number (or less) of exchange interactions.
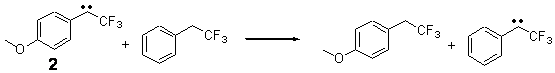
In this recent paper in Angewandte Chemie International Edition Shaik and co-workers apply their EER principle on H-abstraction reactions, and show how dramatic axial ligand effects can be explained by it. The systems under study are [(Cz)(X)MnVO] (Cz: corrolazinato3-, X=None, F-, CN-) complexes (see Figure below), which probably have a singlet ground state (X=None) or a triplet ground state (X=F-, CN-).

More important than the ground state of the reactant is however the spin state of the transition states (TSs). The hydrogen abstraction involves a proton-coupled electron transfer (PCET): the migrating H· radical transfers its radical to the d-block of the metal, while at the same time the proton makes the O-H bond. For the singlet state, one obtains at the TS an open-shell singlet with an alpha electron on the metal and a beta electron on the substrate. In the exchange-enhanced triplet state, there are now three alpha electrons on the metal (with favorable exchange interactions) that gives the dramatic decrease in barrier (from 32 kcal·mol-1 for the singlet to 23.0 kcal·mol-1 for the triplet).
The axial ligand effect has two origins: the exchange interactions become stronger (11.1, 12.4 and 12.9 kcal·mol-1 for the three complexes) while at the same time the d-orbitals become closer in energy (smaller excitation energy).
References
(1) S. Shaik, H. Chen, D. Janardanan, "Exchange-enhanced reactivity in bond activation by metal-oxo enzymes and synthetic reagents", Nature Chem. 2011, 3, 19-27, DOI: 10.1038/NChem.943

This work is licensed under a Creative Commons Attribution 3.0 Unported License.
In a Perspective in Nature Chemistry last year1 Sason Shaik and co-workers described bond activation by metal-oxo enzymes and synthetic reagents. In it, they argued that Hund's rule of maximum multiplicity (valid for atoms) has an analogue for reactions and kinetics of (bio)inorganic species: the exchange-enhanced reactivity (EER). Pathways that increase the number of unpaired and spin-identical electrons on a metal center will be favored by exchange interactions, and hence are favored over pathways that keep the same number (or less) of exchange interactions.
In this recent paper in Angewandte Chemie International Edition Shaik and co-workers apply their EER principle on H-abstraction reactions, and show how dramatic axial ligand effects can be explained by it. The systems under study are [(Cz)(X)MnVO] (Cz: corrolazinato3-, X=None, F-, CN-) complexes (see Figure below), which probably have a singlet ground state (X=None) or a triplet ground state (X=F-, CN-).
More important than the ground state of the reactant is however the spin state of the transition states (TSs). The hydrogen abstraction involves a proton-coupled electron transfer (PCET): the migrating H· radical transfers its radical to the d-block of the metal, while at the same time the proton makes the O-H bond. For the singlet state, one obtains at the TS an open-shell singlet with an alpha electron on the metal and a beta electron on the substrate. In the exchange-enhanced triplet state, there are now three alpha electrons on the metal (with favorable exchange interactions) that gives the dramatic decrease in barrier (from 32 kcal·mol-1 for the singlet to 23.0 kcal·mol-1 for the triplet).
The axial ligand effect has two origins: the exchange interactions become stronger (11.1, 12.4 and 12.9 kcal·mol-1 for the three complexes) while at the same time the d-orbitals become closer in energy (smaller excitation energy).
References
(1) S. Shaik, H. Chen, D. Janardanan, "Exchange-enhanced reactivity in bond activation by metal-oxo enzymes and synthetic reagents", Nature Chem. 2011, 3, 19-27, DOI: 10.1038/NChem.943
This work is licensed under a Creative Commons Attribution 3.0 Unported License.