Chemical Science 2015, early-view (Open-Access)
Contributed by Marcel Swart
The determination of oxidation states of metals in biological systems is not an easy task and can lead to surprises or controversies. This was for instance shown last year when an unusual molybdenum(III) oxidation state in the nitrogenase enzyme was reported [1], or the year before [2] for a Sc-capped iron-oxygen complex where the crystal structure shows a iron(III) oxidation state unlike the expected iron(IV) state based on solution data [3].
The situation is infinitely more complex when more than one metal ion is present, as is the case in the oxygen evolving complex (OEC) of photosystem II that is studied here through a combination of computational chemistry and spectroscopy. The OEC contains 4 manganese and 1 calcium as the active species to convert water into oxygen. Many studies have been performed on the five stages along the catalytic cycle, often with contradictory conclusions about the geometry, electronic structure, oxidation and protonation states involved.
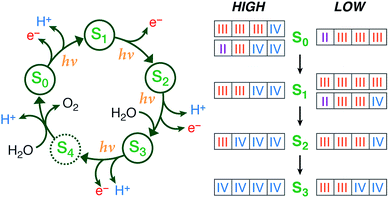 |
Summary of the catalytic cycle with the five stages (reproduced with permission from Chem. Sci., 2015, Advance Article, DOI: 10.1039/C4SC03720K; Published by The Royal Society of Chemistry) |
A breakthrough was a recent atomic resolution crystal structure [4] that largely confirmed the theoretical predictions by Siegbahn [5] about the geometrical positioning of the manganese ions and the oxygens. Still, a detailed understanding of the oxidation states was lacking, which is being explored in the paper by Pantazis and co-workers through a combination of a variety of experimental and theoretical techniques.
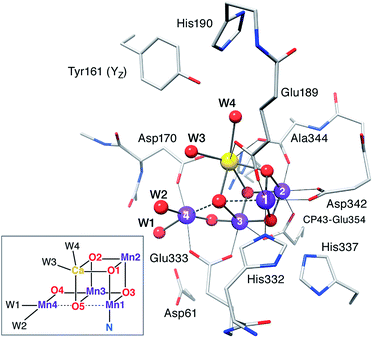 |
Typical model system used in the calculations (reproduced with permission from Chem. Sci., 2015, Advance Article, DOI: 10.1039/C4SC03720K; Published by The Royal Society of Chemistry) |
It is this combination of both theory and experiment, and the systematic exploration of protonation states, open- vs. closed cubane structure, distribution of +3/+4 oxidation states over the different manganese ions that makes this a complete and convincing story for the assignment of high-valent (HV) oxidation states along the catalytic cycle.
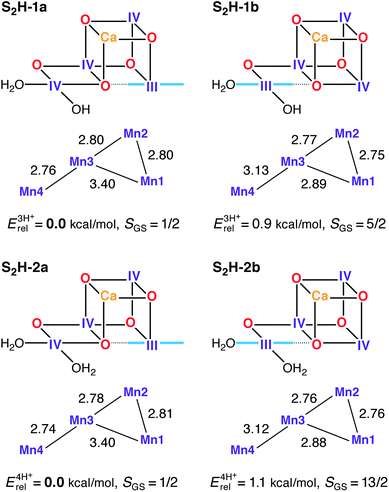 |
One example of the different possible distributions of oxidation states (III or IV) and spin states (1/2, 5/2 or 13/2), for state 2 models (reproduced with permission from Chem. Sci., 2015, Advance Article, DOI: 10.1039/C4SC03720K; Published by The Royal Society of Chemistry) |
The HV assignments made are consistent with the experimental data obtained so far, including a very recent “radiation-damage-free” crystal structure [6], for the first three stages of the catalytic cycle (S1-S3). What remains to be explored is how the enzyme produces the dioxygen molecule in the S4 stage and returns back to the resting state S0. Without any doubt, further unexpected findings will be coming along for this intriguing catalytic cycle.
[1] Chem. Sci. 2014, 5, 3096-3103, DOI: 10.1039/C4SC00337C
[2] Chem. Commun. 2013, 49, 6650-6652, DOI: 10.1039/c3cc42200c
[3] Nature Chem. 2010, 2, 756-759, DOI: 10.1038/nchem.731
[4] Nature 2011, 473, 55-60, DOI: 10.1038/nature09913
[5] Acc. Chem. Res. 2009, 42, 1871-1880, DOI: 10.1021/ar900117k
[6] Nature 2015, 517, 99-103, DOI: 10.1038/nature13991

This work is licensed under a Creative Commons Attribution 3.0 Unported License.