Bickelhaupt, F. M.; Houk, K. N., Angew. Chem. Int. Ed. 2017, 56, 10070-10086
Contributed by Steven Bacharach
Reposted from Computational Organic Chemistry with permission
 '
'
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
Contributed by Steven Bacharach
Reposted from Computational Organic Chemistry with permission
Bickelhaupt and Houk present a nice review of their separately developed, but conceptually identical model for assessing reactivity.1 Houk termed this the “distortion/interaction” model,2 while Bickelhaupt named it “activation strain”.3 The concept is that the activation barrier can be dissected in a distortion or stain energy associated with bringing the reactants into the geometry of the transition state, and the interaction energy is the stabilization energy afforded by the molecular orbital interactions of the reactant components with each other in the transition state.
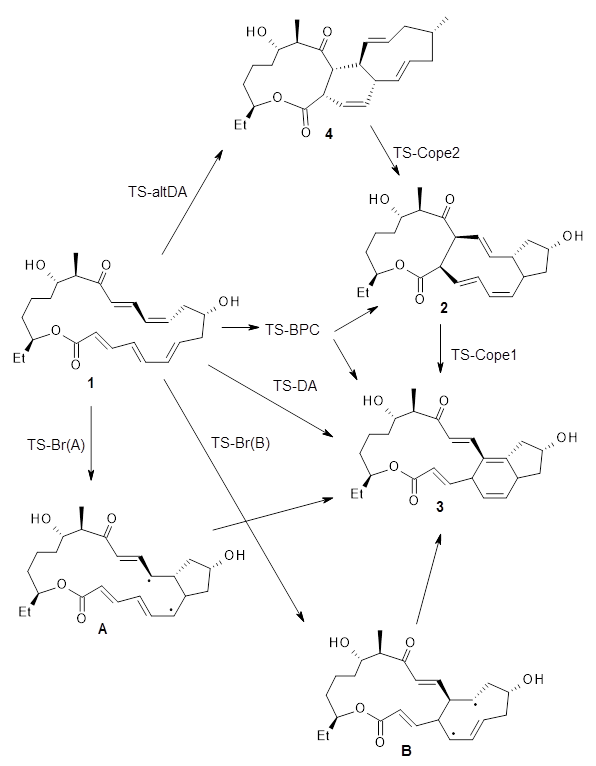
The review discusses a broad range of applications, including SN2 and E2 reactions, pericyclic reactions (including Diels-Alder reactions of enones and the dehdydro Diels-Alder reaction that I have discussed in this blog), a click reaction, a few examples involving catalysis, and the regioselectivity of indolyne (see this post). They also discuss the role of solvent and the relationship of this model to Marcus Theory.
I also want to mention in passing a somewhat related article by Jorgensen and co-authors published in the same issue of Angewandte Chemie as the above review.4 This article discusses the paucity of 10 electron cycloaddition reactions, especially in comparison to the large number of very important cycloaddition reactions involving 6 electrons, such as the Diels-Alder reaction, the Cope rearrangement, and the Claisen rearrangement. While the article does not focus on computational methods, computations have been widely used to discuss 10-electron cycloadditions. The real tie between this paper and the review discussed above is Ken Houk, whose graduate career started with an attempt to perform a [6+4] cycloaddition, and he has revisited the topic multiple times throughout his career.
References
1. Bickelhaupt, F. M.; Houk, K. N., "Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model." Angew. Chem. Int. Ed. 2017, 56, 10070-10086, DOI: 10.1002/anie.201701486.
2. Ess, D. H.; Houk, K. N., "Distortion/Interaction Energy Control of 1,3-Dipolar Cycloaddition Reactivity." J. Am. Chem. Soc. 2007, 129, 10646-10647, DOI: 10.1021/ja0734086
3. Bickelhaupt, F. M., "Understanding reactivity with Kohn-Sham molecular orbital theory: E2-SN2 mechanistic spectrum and other concepts." J. Comput. Chem. 1999, 20, 114-128
4. Palazzo, T. A.; Mose, R.; Jørgensen, K. A., "Cycloaddition Reactions: Why Is It So
Challenging To Move from Six to Ten Electrons?" Angew. Chem. Int. Ed. 2017, 56, 10033-10038, DOI: 10.1002/anie.201701085.
Challenging To Move from Six to Ten Electrons?" Angew. Chem. Int. Ed. 2017, 56, 10033-10038, DOI: 10.1002/anie.201701085.
'This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.