Haichen Li, Christopher Collins, Matteus Tanha, Geoffrey J. Gordon, David J. Yaron (2018)
Highlighted by Jan Jensen

There are increasingly many papers on predicting the molecular energy and other properties using machine learning (ML). Most, if not all, use some similarity measure of the molecular structure to structures in the training set when training. This paper uses DFTB Hamiltonian matrix elements instead and treats the short-range matrix elements as adjustable parameters (weights) to be trained. To make this happen, DFTB is implemented as a layer for deep learning, using the TensorFlow deep learning framework, by recasting the DFTB equations in terms of tensor operations. In this way domain knowledge is incorporated into the ML model. Since the starting values are the "conventional" DFTB parameters one can also view this as refining the DFTB method.
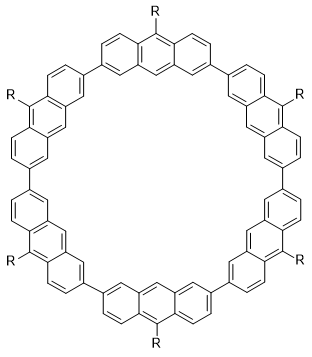
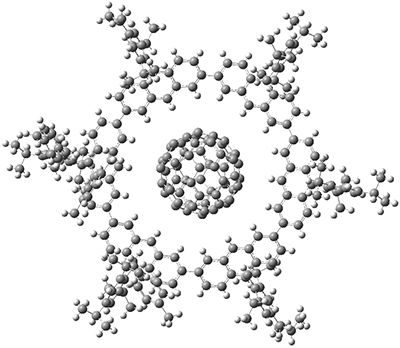
This DFTB-ML approach is evaluated on 15,700 hydrocarbons by comparing the RMSE in energy per heavy atom (Eatom) relative to ωB97X/6-31G(d) reference values. Training on up to 7 heavy atoms and testing on 8 heavy atoms, leads to RMS errors in Eatom of 0.72 kcal/mol, compared to 1.80 using conventional DFTB. Training on up to 4 heavy atoms gives an Eatom RMSE of 1.08 kcal/mol. The results can be further improved by using neural networks to allow the matrix elements to depend on the molecular environment of the atoms.
As the authors point out the performance on the training data remained above chemical accuracy (0.5 kcal/mol) for the total molecular energy, but they offer several interesting ideas on how to improve the performance.

This work is licensed under a Creative Commons Attribution 4.0 International License.
Highlighted by Jan Jensen

There are increasingly many papers on predicting the molecular energy and other properties using machine learning (ML). Most, if not all, use some similarity measure of the molecular structure to structures in the training set when training. This paper uses DFTB Hamiltonian matrix elements instead and treats the short-range matrix elements as adjustable parameters (weights) to be trained. To make this happen, DFTB is implemented as a layer for deep learning, using the TensorFlow deep learning framework, by recasting the DFTB equations in terms of tensor operations. In this way domain knowledge is incorporated into the ML model. Since the starting values are the "conventional" DFTB parameters one can also view this as refining the DFTB method.
This DFTB-ML approach is evaluated on 15,700 hydrocarbons by comparing the RMSE in energy per heavy atom (Eatom) relative to ωB97X/6-31G(d) reference values. Training on up to 7 heavy atoms and testing on 8 heavy atoms, leads to RMS errors in Eatom of 0.72 kcal/mol, compared to 1.80 using conventional DFTB. Training on up to 4 heavy atoms gives an Eatom RMSE of 1.08 kcal/mol. The results can be further improved by using neural networks to allow the matrix elements to depend on the molecular environment of the atoms.
As the authors point out the performance on the training data remained above chemical accuracy (0.5 kcal/mol) for the total molecular energy, but they offer several interesting ideas on how to improve the performance.
This work is licensed under a Creative Commons Attribution 4.0 International License.