Pierre L. Bhoorasingh, Belinda L. Slakman, Fariba Seyedzadeh Khanshan, Jason Y. Cain, and Richard H. West (2017)
Highlighted by Jan Jensen


This work is licensed under a Creative Commons Attribution 4.0 International License.
Highlighted by Jan Jensen

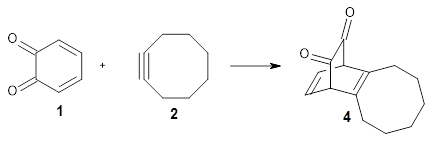
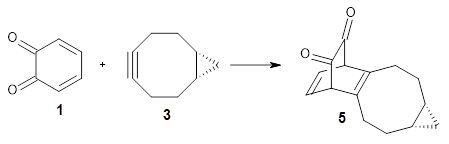
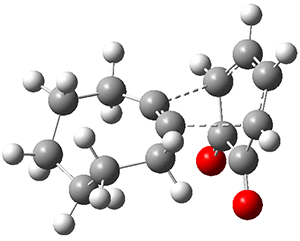
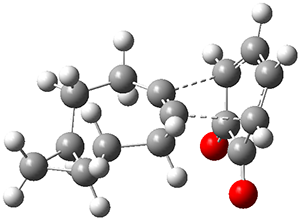
Figure 1 from Bhoorasingh et al. J. Phys. Chem. A 2017, 121, 6896.
Copyright 2017 American Chemical Society
I have written about automated transition state searching before, so I was interested to see how this work differed. Both methods aim at obtaining the best possible guess of the TS structure, which is then used as a starting point for a conventionional TS optimization. In the current work this is done by estimating bond lengths between the reacting atoms using a group contribution method based on known TS structures. These distances are then constrained while a conformational search is performed for the rest of the molecular structure using the UFF force field. The method is described in more detail here.
This approach is thus not too different from the TS template structure approach used in the Schrödinger study, but goes on to perform a conformational search for the TS, which the Schrödinger study did not. So it indeed encouraging to see that the conformational search seems to work and give reasonable results.
Both approaches requires that the atom orders are the same in the reactants and products. In general this is a hard problem and the Schrödinger paper offers one approach to this. However, in the current study the products are automatically generated from the reactants using the Reaction Mechanism Generator (RMG) program in such a way (I believe) that the atom order is preserved.
So if you're interested in a particular TS the current approach is unlikely to be useful since it is rather intimately tied to the RMG program and certain types of chemical reactions. However, if you are interested in these types of chemical reactions then the approach seems quite useful since the entire process is automated and appears quite robust.
More importantly is an important proof-of-concept of what is possible in terms of automation given a large an carefully constructed training set of chemical reactions.
This work is licensed under a Creative Commons Attribution 4.0 International License.