Ermanis, K.; Parkes, K. E. B.; Agback, T.; Goodman, J. M. Org. Biomol. Chem., 2016, 14, 3943-3949
Contributed by Steven Bachrach
Reposted from Computational Organic Chemistry with permission
 '
'
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
Computational chemistry has had a remarkable impact on the field of structure determination by NMR spectroscopy. The ability to efficiently compute 13C and 1H chemical shifts allows for comparison of the computed chemical shifts of potential structures against the experimental values, a tremendous aid in structure determination (see some examples in previous posts). Goodman and Smith developed the DP4 method1 (see this post) to assist in identifying proper structures by means of statistical distribution of errors and Bayes Theorem.
The Goodman group now reports on workflow solutions to structure prediction using DP4.2 They explore the use of open source computational tools both for predicting conformations and for computing the chemical shifts. They use a set of 10 drugs to test the performance. In general, the original DP4 method works very well in predicting drug structure, despite the fact that DP4 parameters were developed for natural products. The only failure is for simvastatin, where the large number of diastereomers and conformational flexibility prove to be too complex. The open source tools perform just slightly less effectively than the commercial packages, but are certainly a viable route for those with limited resources. The authors also provide a series of python scripts that allow users to create a seamless workflow; these should prove most helpful to the structure determination community.
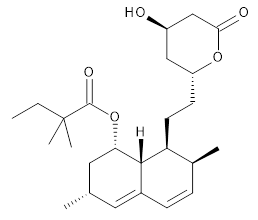 Simvastatin |
References
1) Smith, S. G.; Goodman, J. M. "Assigning Stereochemistry to Single Diastereoisomers by GIAO
NMR Calculation: The DP4 Probability," J. Am. Chem. Soc. 2010, 132, 12946-12959, DOI:10.1021/ja105035r.
NMR Calculation: The DP4 Probability," J. Am. Chem. Soc. 2010, 132, 12946-12959, DOI:10.1021/ja105035r.
2) Ermanis, K.; Parkes, K. E. B.; Agback, T.; Goodman, J. M. “Expanding DP4: application to drug compounds and automation,” Org. Biomol. Chem., 2016, 14, 3943-3949, DOI: 10.1039/c6ob00015k.
InChIs
Simvastatin: InChI=1S/C25H38O5/c1-6-25(4,5)24(28)30-21-12-15(2)11-17-8-7-16(3)20(23(17)21)10-9-19-13-18(26)14-22(27)29-19/h7-8,11,15-16,18-21,23,26H,6,9-10,12-14H2,1-5H3/t15-,16-,18+,19+,20-,21-,23-/m0/s1
InChIKey=RYMZZMVNJRMUDD-HGQWONQESA-N
InChIKey=RYMZZMVNJRMUDD-HGQWONQESA-N
'This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
No comments:
Post a Comment