Buevich, A. V. J. Org. Chem. 2016, 81, 485–501
Contributed by Steven Bachrach
Reposted from Computational Organic Chemistry with permission
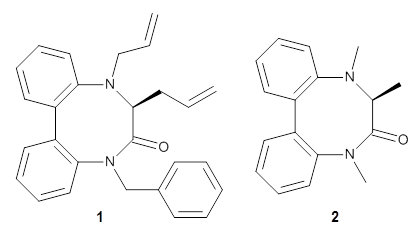
Atropisomers are stereoisomer that differ by axial symmetry, such as in substituted biphenyls or allenes. These acyclic systems have received a fair amount of attention, but now Buevich has looked at atropisomerization that occurs in a ring system.1 1 has a biphenyl as part of the eight-member ring, and the biphenyl can exist in either an M or P orientation. Since C3 is chiral (S), the two isomers are (M,S)-1and (P,S)-1. Variable temperature NMR analysis concludes that (P,S)-1 is 1.19 kcal mol-1 more stable than (M,S)-1, and the barrier for the interchange (P,S)-1 → (M,S)-1 is 26.77 kcal mol-1.

This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
Contributed by Steven Bachrach
Reposted from Computational Organic Chemistry with permission
Atropisomers are stereoisomer that differ by axial symmetry, such as in substituted biphenyls or allenes. These acyclic systems have received a fair amount of attention, but now Buevich has looked at atropisomerization that occurs in a ring system.1 1 has a biphenyl as part of the eight-member ring, and the biphenyl can exist in either an M or P orientation. Since C3 is chiral (S), the two isomers are (M,S)-1and (P,S)-1. Variable temperature NMR analysis concludes that (P,S)-1 is 1.19 kcal mol-1 more stable than (M,S)-1, and the barrier for the interchange (P,S)-1 → (M,S)-1 is 26.77 kcal mol-1.
To identify the process for this atropisomerization process, he utilized B3LYP/6-31G(d) computations of the model system 2. A variety of different techniques were used to identify the local energy minimum conformations of both (M,S)-2 and (P,S)-2, and the lowest energy conformers (M1 for (P,S)-2 and M4 for (M,S)-2) are shown in Figure 1. He then produced a series of 2-D potential energy surfaces varying two of the dihedral angles defining the eight-member ring to help identify potential initial geometries for searching for transition states. (As an aside, this procedure ended up identifying a few additional local energy minima not identified in the initial conformational search – and these all have trans amide groups instead of the cis relationship found initially. These trans isomer are considerably higher in energy than the conformers.) With this model and this computational level, (P,S)-2 is 0.76 kcal mol-1 lower in energy than (M,S)-2.
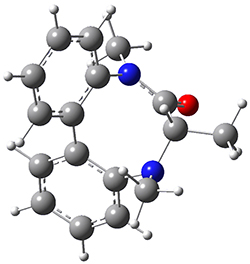
M1
0.0 | 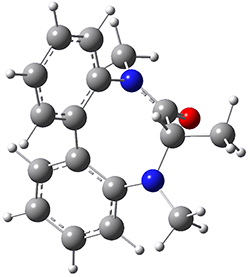 TS1 3.54 |
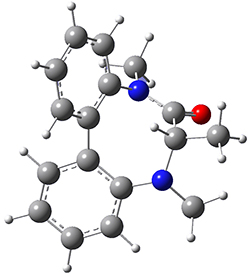 M2 2.21 | 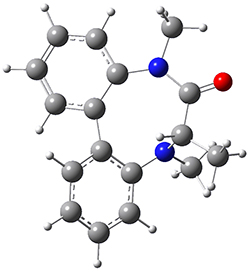 TS4 25.83 |
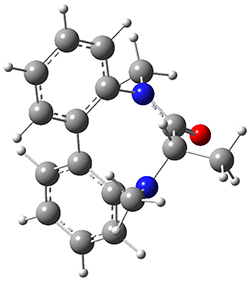 M4 0.76 |
Table 1. B3LYP/6-31G(d) optimized geometries and relative free energies of some critical points along the lowest energy pathway taking (P,S)-2 → (M,S)-2.
A number of transition states were identified, and the lowest energy pathway that takes M1 into M4 first crosses TS1 to make the minimum M2, which than passes a high barrier (25.8 kcal mol-1) to go to M4. This barrier is in reasonable agreement with the experimental barrier for 1. These TSs are also shown in Figure 1.
Buevich analyzes the conformational process by examination of the changes in the ring dihedral angles following this reaction path. As expected, crossing the highest barrier requires a combination of torsional rotations, but essentially one at a time moving clockwise about the ring.
References
(1) Buevich, A. V. "Atropisomerization of 8-Membered Dibenzolactam: Experimental NMR and Theoretical DFT Study," J. Org. Chem. 2016, 81, 485–501 DOI: 10.1021/acs.joc.5b02321.
InChIs
1: InChI=1S/C27H26N2O/c1-3-12-26-27(30)29(20-21-13-6-5-7-14-21)25-18-11-9-16-23(25)22-15-8-10-17-24(22)28(26)19-4-2/h3-11,13-18,26H,1-2,12,19-20H2/t26-/m0/s1
InChIKey=IYYACMIVKDJSJD-SANMLTNESA-N
InChIKey=IYYACMIVKDJSJD-SANMLTNESA-N
2: InChI=1S/C17H18N2O/c1-12-17(20)19(3)16-11-7-5-9-14(16)13-8-4-6-10-15(13)18(12)2/h4-12H,1-3H3/t12-/m0/s1
InChIKey=NIBPKKMKNFOPRM-LBPRGKRZSA-N
InChIKey=NIBPKKMKNFOPRM-LBPRGKRZSA-N
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
No comments:
Post a Comment