Kwan, E. E.; Liu, R. Y. J. Chem. Theor. Comput. 2015, 11, 5083-5089
Contributed by Steven Bachrach
Reposted from Computational Organic Chemistry with permission
[Editors note: this paper has also been highlighted by Jan Jensen]

This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
Contributed by Steven Bachrach
Reposted from Computational Organic Chemistry with permission
The prediction of NMR chemical shifts and coupling constants through ab initio computation is a major development of the past decade in computational organic chemistry. I have written about many developments on this blog. An oft-used method is a linear scaling of the computed chemical shifts to match those of some test set. Kwan and Liu wondered if the dynamics of molecular motions might be why we need this correction.1
They suggest that the chemical shift can be computed as
<σ> = σ(static molecule using high level computation) + error
where the error is the obtained by using a low level computation taking the difference between the chemical shifts obtained on a dynamic molecule less that obtained with a static molecule. The dynamic system is obtained by performing molecular dynamics of the molecule, following 25 trajectories and sampling every eighth point.
They find outstanding agreement for the proton chemical shift of 12 simple molecules (mean error of 0.02 ppm) and the carbon chemical shift of 19 simple molecules (mean error of 0.5 ppm) without any scaling. Similar excellent agreement is found for a test set of natural products.
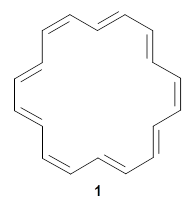
They finish up with a discussion of [18]annulene 1. The structure of 1 is controversial. X-ray crystallography indicates a near D6h geometry, but the computed NMR shifts using a D6h geometry are in dramatic disagreement with the experimental values, leading Schleyer to suggest a C2 geometry. Kwan and Liu applied their dynamic NMR method to the D6h, D3h, and C2 structures, and find the best agreement with the experimental chemical shifts are from the dynamic NMR initiated from the D6h geometry. Dynamic effects thus make up for the gross error found with the static geometry, and now bring the experimental and computational data into accord.
One final note on this paper. The authors indicate that they have filed a provisional patent on their method. I am disturbed by this concept of patenting a computational methodology, especially in light of the fact that many other methods have been made available to the world without any legal restriction. For example, full details including scripts to apply Tantillo’s correction method are available through the Cheshire site and a web app to implement Goodman’s DP4 method are available for free. Provisional patents are not available for review from the US Patent Office so I cannot assess just what is being protected here. However, I believe that this action poses a real concern over the free and ready exchange of computational methodologies and ideas.
References
(1) Kwan, E. E.; Liu, R. Y. "Enhancing NMR Prediction for Organic Compounds Using Molecular Dynamics," J. Chem. Theor. Comput. 2015, 11, 5083-5089, DOI: 10.1021/acs.jctc.5b00856.
InChIs
1: InChI=1S/C18H18/c1-2-4-6-8-10-12-14-16-18-17-15-13-11-9-7-5-3-1/h1-18H/b2-1-,3-1+,4-2+,5-3+,6-4+,7-5-,8-6-,9-7+,10-8+,11-9+,12-10+,13-11-,14-12-,15-13+,16-14+,17-15+,18-16+,18-17-
InChIKey=STQWAGYDANTDNA-DWSNDWDZSA-N
InChIKey=STQWAGYDANTDNA-DWSNDWDZSA-N
[Editors note: this paper has also been highlighted by Jan Jensen]
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
No comments:
Post a Comment