Dimitrios G. Liakos, Manuel Sparta, Manoj K. Kesharwani, Jan M. L. Martin, and Frank Neese J. Chem. Theory Comput. 2015, 11, 1525−1539
Contributed by +Jan Jensen

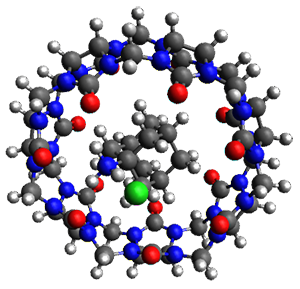
Two years ago Neese and co-workers published the first CCSD(T) calculation on a protein (crambin), which, according to this latest paper remains "the largest coupled cluster calculations reported to date". The calculations were made possible through the domain based local pair natural orbital (DLPNO) implementation of CCSD(T), which includes a variety of approximations with associated thresholds that control the accuracy.
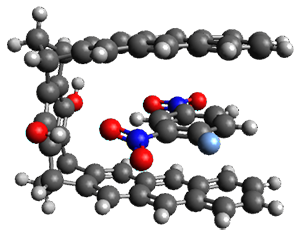
The study includes molecules up to 30 atoms including a "real-life" organometallic catalysis and a study of 51 conformers of melatonin. Here the focus is very much on accuracy and little or no information was given on computational cost. So I encourage any reader with practical experience using this method to leave a comment with their experiences.
Accurate CCSD(T) benchmark data sets for small complexes has been incredibly useful in parameterizing dispersion corrections and semi-empirical methods. It looks like DLPNO-CCSD(T) has made it practical to construct similar benchmark sets for chemical reactivity of organometallic and bio-catalytic models systems which currently are too big to be treated with conventional CCSD(T).
Contributed by +Jan Jensen
Reprinted with permission from J. Chem. Theory Comput. 2015, 11, 1525-1539.
Copyright (2015) American Chemical Society.
Copyright (2015) American Chemical Society.
Two years ago Neese and co-workers published the first CCSD(T) calculation on a protein (crambin), which, according to this latest paper remains "the largest coupled cluster calculations reported to date". The calculations were made possible through the domain based local pair natural orbital (DLPNO) implementation of CCSD(T), which includes a variety of approximations with associated thresholds that control the accuracy.
The current paper defines three default thresholds termed "LoosePNO", "NormalPNO", "and TightPNO" recommended for rapid estimates, general thermochemistry and kinetics, and non-covalent interactions and conformational equilibria, respectively. The latter two provide relative energies with 1 kcal/mol of conventional CCSD(T) calculations.
The study includes molecules up to 30 atoms including a "real-life" organometallic catalysis and a study of 51 conformers of melatonin. Here the focus is very much on accuracy and little or no information was given on computational cost. So I encourage any reader with practical experience using this method to leave a comment with their experiences.
Accurate CCSD(T) benchmark data sets for small complexes has been incredibly useful in parameterizing dispersion corrections and semi-empirical methods. It looks like DLPNO-CCSD(T) has made it practical to construct similar benchmark sets for chemical reactivity of organometallic and bio-catalytic models systems which currently are too big to be treated with conventional CCSD(T).