Marell, D. J.; Furan, L. R.; Woods, B. P.; Lei, X.; Bendelsmith, A. J.; Cramer, C. J.; Hoye, T. R.; Kuwata, K. T. J. Org. Chem. 2015 ASAP
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission

This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission
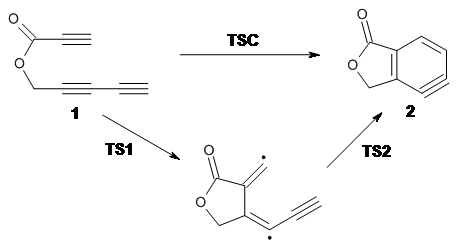
Cramer, Hoye, Kuwata and coworkers have examined the intramolecular cyclization of an alkyne with a diyne.1 Their model system is 1, which can cyclize through a concerted transition state TSC togive the benzyne product 2, or it can proceed through a stepwise pathway, first going through TS1 to form the intermediate INT¸ before traversing through a second transition state TS2 and on to product 2. Using both computations and experiments, they examined a series of compounds with differing substituents at the ends of the two yne moieties.
The experiments show almost the exact same rate of reaction regardless of the terminal substituents. This includes the parent case where the terminal substituents are hydrogens and also the case where the terminal substituents (which end up on adjacent centers on the benzyne ring) are bulky TMS groups. And though there is no real rate effect due to changes in solvent or the presence of light or triplet oxygen, which suggest a concerted reaction, these substituent effects argue for a step wise reaction.
SMD(o-dichlorobenzene)/B3LYP-D3BJ/6-311+G-(d,p)//M06-2X/6-311+G(d,p) computations help explain these observations. Shown in Figure 1 are the optimized geometries and relative energies of the critical points on the reaction surface for the conversion of 1 into 2, and these results are similar with the other substituents as well.
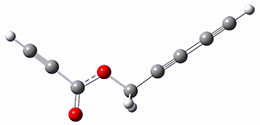
1
(0.0) | 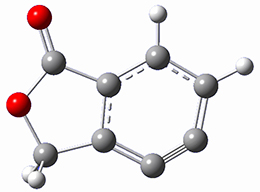
2
(-56.9) |
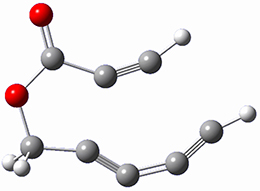
TSC
(31.5) | |
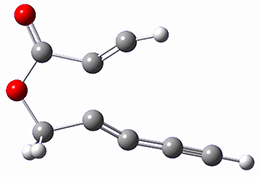
TS1
(25.5) | 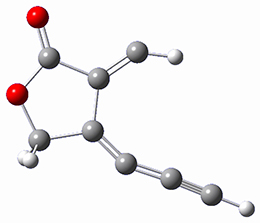
INT
(18.8) |
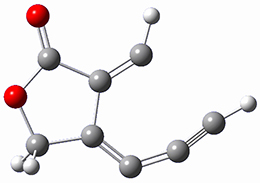
TS2
(18.1) |
Figure 1. SMD(o-dichlorobenzene)/B3LYP-D3BJ/6-311+G-(d,p)//M06-2X/6-311+G(d,p) optimized geometries and relative energies (kcal mol-1).
The first thing to note is that the concerted TSC is higher in energy than the stepwise TS1. The wavefunction for TSC unstable towards moving from a restricted to unrestricted formalism. Reoptimization of some of these concerted TSs actually led to the stepwise TS.
The next item of note is that TS2 for this case is actually lower in energy than the intermediate (this is a true TS on the energy surface, but when zero-point energy and other thermal corrections are included, it becomes lower in energy than INT). For some of the cases the second TS lies above the intermediate, but typically by a small amount.
Therefore, the mechanism of this reaction is stepwise, but the second step might have such a small barrier (or even no barrier) that one might consider this to be concerted, though highly asymmetric and really bearing little resemblance to more traditional concerted pericyclic reactions.
The authors obliquely hinted at some potential interesting dynamics. I suspect that molecular dynamics calculations will show no effect of that second TS, and one might observe some interesting dynamics, which could be seen in kinetic isotope experiments.
References
(1) Marell, D. J.; Furan, L. R.; Woods, B. P.; Lei, X.; Bendelsmith, A. J.; Cramer, C. J.; Hoye, T. R.; Kuwata, K. T. "Mechanism of the Intramolecular Hexadehydro-Diels–Alder Reaction," J. Org. Chem. 2015 ASAP, DOI:10.1021/acs.joc.5b01356.
InChIs
1: InChI=1S/C8H4O2/c1-3-5-6-7-10-8(9)4-2/h1-2H,7H2
InChIKey=MGXDIFXPYGGQLF-UHFFFAOYSA-N
InChIKey=MGXDIFXPYGGQLF-UHFFFAOYSA-N
2: InChI=1S/C8H4O2/c9-8-7-4-2-1-3-6(7)5-10-8/h2,4H,5H2
InChIKey=MYFORDRJCVOBTH-UHFFFAOYSA-N
InChIKey=MYFORDRJCVOBTH-UHFFFAOYSA-N
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
No comments:
Post a Comment