Antony, J.; Sure, R.; Grimme, S. Chem. Commun. 2015, 51, 1764-1774
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission

This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission
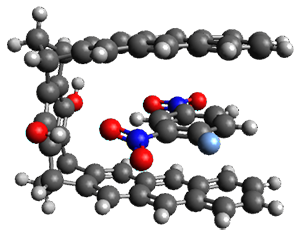
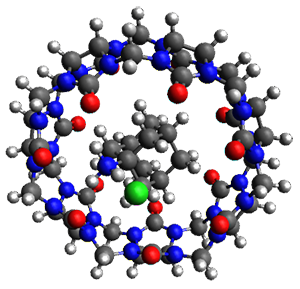
Grimme and coworkers have a featured article on computing host-guest complexes in a recentChemComm.1 They review the techniques his group has pioneered, particularly dispersion corrections for DFT and ways to treat the thermodynamics in moving from electronic energy to free energy. they briefly review some studies done by other groups. They conclude with a new study of eight different host guest complexes, three of which are shown in Figure 1.
1
| 
2
|

3
| |
Figure 1. TPSS-D3(BJ)/def2-TZVP optimized structures of 1-3.
These eight host-guest complexes are fairly large systems, and the computational method employed means some fairly long computations. Geometries were optimized at TPSS-D3(BJ)/def2-TZVP, then single point energy determined at PW6B95-D3(BJ)/def2-QZVP. Solvent was included using COSMO-RS. The curcurbituril complex 2 includes a counterion (chloride) along with the guest adamantan-1-aminium. Overall agreement of the computed free energy of binding with the experimental values was very good, except for 3 and the related complex having a larger nanohoop around the fullerene. The error is due to problems in treating the solvent effect, which remains an area of real computational need.
An interesting result uncovered is that the binding energy due to dispersion is greater than the non-dispersion energy for all of these complexes, including the examples that are charged or where hydrogen bonding may be playing a role in the bonding. This points to the absolute necessity of including a dispersion correction when treating a host-guest complex with DFT.
As an aside, you’ll note one of the reasons I was interested in this paper: 3 is closely related to the structure that graces the cover of the second edition of my book.
References
(1) Antony, J.; Sure, R.; Grimme, S. "Using dispersion-corrected density functional theory to understand supramolecular binding thermodynamics," Chem. Commun. 2015, 51, 1764-1774, DOI:10.1039/C4CC06722C.
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
No comments:
Post a Comment