Karmakar, S.; Datta, A. Angew. Chem. Int. Ed. 2014, 53, 9587-9591
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission

This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission
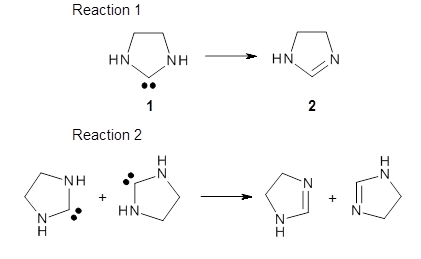
Organic chemists are beginning to recognize that tunneling may be more pervasive than previously thought. This blog has noted a number of interesting occurrences of tunneling, and here’s one more, by Karmakar and Datta.1
The barrier for the intramolecular earrangement (Reaction 1) taking the carbene 1 into 2 is estimated to be 44.1 kcal mol-1 at M06-2X/6-31+G(d,p), prohibitively large. However, the intermolecular rearrangement (Reaction 2) has a much smaller barrier of 11.4 kcal mol-1. The structures of the transition states for these two reactions are shown in Figure 1.
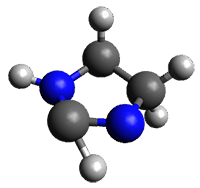
TSintra
| 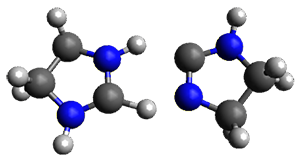
TSinter
|
Figure 1. M06-2X/6-31+G(d,p) optimized transition states for Reactions 1 and 2.
Given that the barrier width is likely to be very small for the intramolecular route, perhaps tunneling may play a role. The rate predicted with canonical variational transition-state theory (CVT) and small curvature tunneling (SCT) at 298K is negligible. However, for the intermolecular process, the rate at 298K including tunneling is 3.56 x 104 s-1, more than 10 times great than predicted with CVT alone, and tunneling makes a dramatically larger difference at lower temperatures.
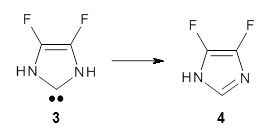
The intermolecular barrier for the rearrangement of 3 into 4 is very small, only 1.6 kcal mol-1.
This manifests in a very interesting rate prediction: the reaction is actually predicted to be slower at temperatures above 150K when tunneling is included than when tunneling is omitted. This is a result of quantum mechanical reflection off of the barrier, and this becomes noticeable with the very small barrier. In addition, the kinetic isotope effects are smaller than expected when D is substituted in for H. These predictions await experimental confirmation.
This manifests in a very interesting rate prediction: the reaction is actually predicted to be slower at temperatures above 150K when tunneling is included than when tunneling is omitted. This is a result of quantum mechanical reflection off of the barrier, and this becomes noticeable with the very small barrier. In addition, the kinetic isotope effects are smaller than expected when D is substituted in for H. These predictions await experimental confirmation.
References
(1) Karmakar, S.; Datta, A. "Tunneling Assists the 1,2-Hydrogen Shift in N-Heterocyclic Carbenes," Angew. Chem. Int. Ed. 2014, 53, 9587-9591, DOI: 10.1002/anie.201404368.
InChIs:
1: InChI=1S/C3H6N2/c1-2-5-3-4-1/h4-5H,1-2H2
InChIKey=JKQUEGZDRZXJNY-UHFFFAOYSA-N
InChIKey=JKQUEGZDRZXJNY-UHFFFAOYSA-N
2: InChI=1S/C3H6N2/c1-2-5-3-4-1/h3H,1-2H2,(H,4,5)
InChIKey=MTNDZQHUAFNZQY-UHFFFAOYSA-N
InChIKey=MTNDZQHUAFNZQY-UHFFFAOYSA-N
3: InChI=1S/C3H2F2N2/c4-2-3(5)7-1-6-2/h6-7H
InChIKey=LHUPDFSUHVZFPD-UHFFFAOYSA-N
InChIKey=LHUPDFSUHVZFPD-UHFFFAOYSA-N
4: InChI=1S/C3H2F2N2/c4-2-3(5)7-1-6-2/h1H,(H,6,7)
InChIKey=KXXZDIFMEWOLPE-UHFFFAOYSA-N
InChIKey=KXXZDIFMEWOLPE-UHFFFAOYSA-N
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
No comments:
Post a Comment