Chen, B.; Hrovat, D. A.; West, R.; Deng, S. H. M.; Wang, X.-B.; Borden, W. T. J. Am. Chem. Soc. 2014, 136, 12345-12354
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission

This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission
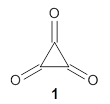
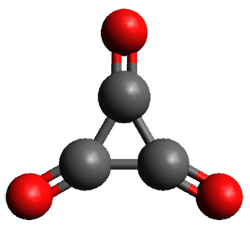
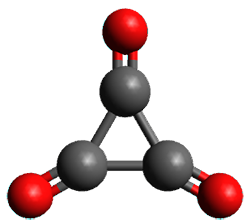
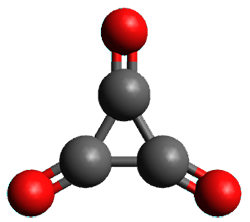
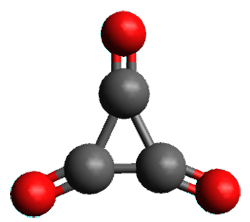
As recently explicated by Wang and Borden using NIPE spectroscopy and computations, the potential energy surface of cyclopropyl-1,2,3-trione 1 is remarkably complex.1 (U)CCSD(T)//aug-cc-pVTZ computations of the D3h singlet (the 1A1’ state shown in Figure 1) is actually a hilltop, possessing two imaginary frequencies. Distorting the structure as indicated by these imaginary frequencies and then optimizing the structure leads directly to dissociation to three CO molecules. Thus, (CO)3 does not exist as a stable minima on the singlet surface.
The D3h triplet (the 3E” state shown in Figure 1) is not a critical point on the surface; due to the Jahn-Teller effect is distorts into two different states: the 3B1 state which is a local energy minimum, and the 3A2 state which is a transition state between the symmetry-related 3B1 states.
So, this implies the possibility of a very interesting NIPE experiment. If the radical anion (CO)3-.
loses an electron and goes to the singlet surface, it lands at a hilltop(!) and should have a very short lifetime. If it goes to the triplet surface, it lands at either a transition state (3A2) and again should have a short lifetime, or it can land at the 3B1 state and perhaps have some lifetime before it dissociates by losing one CO molecule.
loses an electron and goes to the singlet surface, it lands at a hilltop(!) and should have a very short lifetime. If it goes to the triplet surface, it lands at either a transition state (3A2) and again should have a short lifetime, or it can land at the 3B1 state and perhaps have some lifetime before it dissociates by losing one CO molecule.
1-.
| |
1A1’
| 
3E”
|
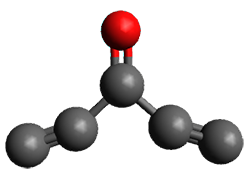
3B1
| 
3A2
|
Figure 1. (U)CCSD(T)//aug-cc-pVTZ optimized geometries of 1 and its radical anion.
The NIPE spectrum identifies three transitions. By comparing the energies of the electron loss seen in the experiment with the computations, along with calculating the Franck-Condon factors using the computed geometries and vibrational frequencies, the lowest energy transition is to the 1A1’ state, and the second transition is part of the vibrational progression also to the 1A1’ state. This is the first identification of vibrational frequencies associated with a hilltop structure. The third transition is to the 3A2 state. No transition to the 3B1 state is found due to the large geometric difference between the radical anion and the3B1 state; the Franck-Condon factors are zero due to no overlap of their wavefunctions.
Once again, the power of the symbiotic relationship between experiment and computation is amply demonstrated in this paper.
References
(1) Chen, B.; Hrovat, D. A.; West, R.; Deng, S. H. M.; Wang, X.-B.; Borden, W. T. "The Negative Ion Photoelectron Spectrum of Cyclopropane-1,2,3-Trione Radical Anion, (CO)3•– — A Joint Experimental and Computational Study," J. Am. Chem. Soc. 2014, 136, 12345-12354, DOI: 10.1021/ja505582k.
InChIs
1: InChI=1S/C3O3/c4-1-2(5)3(1)6
InChIKey=RONYDRNIQQTADL-UHFFFAOYSA-N
InChIKey=RONYDRNIQQTADL-UHFFFAOYSA-N
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
No comments:
Post a Comment