(1) Jahn, M. K.; Dewald, D.; Vallejo-López, M.; Cocinero, E. J.; Lesarri, A.; Grabow, J.-U. "Rotational Spectra of Bicyclic Decanes: The Trans Conformation of (-)-Lupinine," J. Phys. Chem. A 2013
(2) Shepherd, D. J.; Broadwith, P. A.; Dyson, B. S.; Paton, R. S.; Burton, J. W. "Structure Reassignment of Laurefurenynes A and B by Computation and Total Synthesis," Chem. Eur. J. 2013, 19, 12644-12648
1
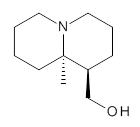
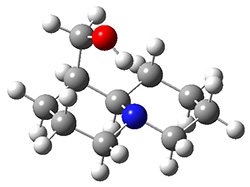
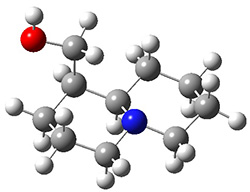
The lowest energy structures had the expected trans ring fusion, with a trans relationship between the hydrogen on the bridgehead carbon (C9) and the hydroxymethyl group. This corresponds to either the (R,R) or (S,S) isomer. The three lowest energy structures are shown in Figure 1. Unfortunately, the geometry for the lowest energy isomer provided in the Supporting Materials is wrong, and the authors did not supply the geometries of the other isomers. This situation is unacceptable! Reviewers and editors must do a better job in policing the Supporting Materials; there is no excuse for not including all of the optimized structures, and better yet, in a more usable format that what has been done here. I have reoptimized these structures at M06-2x/6-31G(d). The lowest energy conformer 1a does possess the expected internal hydrogen bond.

This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
(2) Shepherd, D. J.; Broadwith, P. A.; Dyson, B. S.; Paton, R. S.; Burton, J. W. "Structure Reassignment of Laurefurenynes A and B by Computation and Total Synthesis," Chem. Eur. J. 2013, 19, 12644-12648
I have not discussed any papers that utilize computations to confirm chemical structure in a while, so here are two recent examples.
Grabow has utilized MP2 and M06-2x computations to confirm the lowest energy conformation of (-)-lupinine 1.1 The interesting structural aspect of this compound is the possibility of an intramolecular hydrogen bond linking the hydroxyl group with the amine.
1
Using molecular mechanics, the authors identified 57 structures within 50 kJ mol-1 of each other. These geometries were reoptimized at MP2/6-311++G(d,p) and M06-2x/6-311++G(d,p).
The lowest energy structures had the expected trans ring fusion, with a trans relationship between the hydrogen on the bridgehead carbon (C9) and the hydroxymethyl group. This corresponds to either the (R,R) or (S,S) isomer. The three lowest energy structures are shown in Figure 1. Unfortunately, the geometry for the lowest energy isomer provided in the Supporting Materials is wrong, and the authors did not supply the geometries of the other isomers. This situation is unacceptable! Reviewers and editors must do a better job in policing the Supporting Materials; there is no excuse for not including all of the optimized structures, and better yet, in a more usable format that what has been done here. I have reoptimized these structures at M06-2x/6-31G(d). The lowest energy conformer 1a does possess the expected internal hydrogen bond.
1a
(0.0) | |
1b
(10.4) | 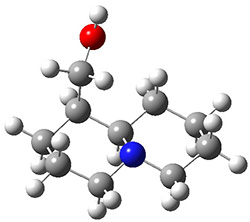
12
(11.5) |
Figure 1. M06-2x/6-31G(d) optimized structures of the three lowest energy conformers of 1, with relative free energies in kJ mol-1.
Table 1 provides a comparison of the MP2 computed values of important structural parameters along with the experimental values obtained from a microwave experiment. The agreement with the computed values for 1a provides strong evidence that this is the structure of (-)-lupinine.
Table 1. Comparison of MP2 and experimental structural parameters of 1.a
Expt. (1)
|
MP2 (1a)
| |
A
|
1414.126
|
1425.8
|
B
|
811.672
|
815.1
|
C
|
671.530
|
677.1
|
ΔJ
|
0.0255
|
0.023
|
ΔJK
|
0.0639
|
0.065
|
ΔK
|
0.0037
|
0.0022
|
χaa
|
1.9973
|
2.0
|
χbb
|
1.062
|
1.1
|
χcc
|
-3.059
|
-3.1
|
aRotational constants (A, B, C) in MHz, centrifugal distortion constants (ΔJ, ΔJK, ΔK) in kHz, and nuclear quadrupole coupling tensor elements (χaa, χbb, χcc) in MHz.
The second study utilizes computed NMR chemical shifts to discriminate potential diastereomeric structures. Laurefurenyne A was first assigned the structure 2 based on 1D and 2D NMR experiments. However, based on potential biochemical analogy to other compounds, Paton and Burton2 had doubts about this structure. In addition to synthesizing the natural material, they performed an extensive computational study of the chemical shifts of the diastereomers. For each of the 32 possible diastereomers, they performed a Monte Carlo search of the conformational space using molecular mechanics. The structures of all isomers within 10 kJ mol-1 of the lowest energy structure were reoptimized at ωB97X-D/6-31G(d) with PCM (CHCl3) and chemical shifts obtained at mPW1PW91/6-311G(d,p). Final chemical shifts were obtained using a Boltzmann weighting. The computed values for 2were quite off from the experimental values, with a mean unsigned error of 1.5 ppm. A better assessment was provided with the DP4 method, which indicated that 3 has the highest probability of being the correct structure, a structure consistent with the likely biosynthetic pathway.
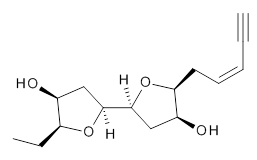 2 | 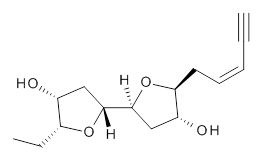 3 |
References
(1) Jahn, M. K.; Dewald, D.; Vallejo-López, M.; Cocinero, E. J.; Lesarri, A.; Grabow, J.-U. "Rotational Spectra of Bicyclic Decanes: The Trans Conformation of (-)-Lupinine," J. Phys. Chem. A 2013, DOI:10.1021/jp407671m.
(2) Shepherd, D. J.; Broadwith, P. A.; Dyson, B. S.; Paton, R. S.; Burton, J. W. "Structure Reassignment of Laurefurenynes A and B by Computation and Total Synthesis," Chem. Eur. J. 2013, 19, 12644-12648, DOI: 10.1002/chem.201302349.
InChIs
(-)-Lupinine 1: InChI=1S/C11H21NO/c1-11-6-2-3-7-12(11)8-4-5-10(11)9-13/h10,13H,2-9H2,1H3/t10-,11+/m0/s1
InChIKey=WVDUAOYJLFVEMW-WDEREUQCSA-N
InChIKey=WVDUAOYJLFVEMW-WDEREUQCSA-N
Laurefurenyne A 3: InChI=1S/C14H20O4.C2H6/c1-3-4-5-6-12-11(16)8-14(18-12)13-7-10(15)9(2)17-13;1-2/h1,4-5,9-16H,6-8H2,2H3;1-2H3/b5-4-;/t9-,10-,11-,12+,13-,14+;/m1./s1
InChIKey=ZYESDGGYHKCHMJ-SKFGUVSTSA-N
InChIKey=ZYESDGGYHKCHMJ-SKFGUVSTSA-N
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
No comments:
Post a Comment