Lane, J. R.; Contreras-García, J.; Piquemal, J.-P.; Miller, B. J.; Kjaergaard, H. G. J. Chem. Theor. Comput. 2013, 9, 3263
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission

This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission
One of the more controversial components of Bader’s Atoms-In-Molecules (AIM) theory is the contention that there is a one-to-one correspondence between the existence of a bond critical point and the existence of a chemical bond. I discuss this matter in my book and also in these posts (1 and 2). Lane and co-workers now examine this relationship with regard to hydrogen bonds.1
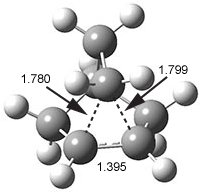
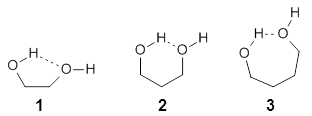
They examine the topological structure of the electron density of the series 1,2-ethanediol 1, 1,3-propanediol 2, and 1,3-butanediol 3. They find a bond critical point (bcp) between the hydrogen of one hydroxyl group and the oxygen of the second hydroxyl group for the two large compounds 2 and 3. This forms a ring, and a ring critical point is located as well. However, for 1 they find no bond critical point associated with what might be the intramolecular hydrogen bond in 1. For all three diols, the OH stretching frequencies are diminished relative to monoalcohols. So geometrically and spectroscopically there appears to be a hydrogen bond, but rigorous application of Bader’s notion of bonding says that there is no “bond” in 1.
Lane and coworkers go on to show that the electron density in the three diols is really topologically identical, just differing in a matter of degree. They conclude that the existence of a bond critical point should not be the sole arbiter of bonding, but one of the criteria that can be utilized to assess bonding.
While I am not at all in conflict with this conclusion, the paper contains some issues that need be addressed. First off, “bonding” is not a concept of either-or, rather there is a continuum of bonding. Hydrogen bonding should not at all be confused with covalent or ionic bonding – it is dramatically weaker and so one might consider whether the bcp criteria is applicable at all. The authors really fall into this trap stating “… the absence of a BCP should not necessarily be considered evidence as to the absence of a chemical bond (emphasis mine).” Do we want to consider a hydrogen bond as a chemical bond?
I think the key element overlooked in this study is the strength of the “hydrogen bond”. While not determined in the study, undoubtedly the hydrogen bond strength increases in the order 1 < 2 < 3. What is really to be gained by arguing there is or is not a “hydrogen bond” in all or some of these three molecules? The ring-like conformation is the lowest energy conformation for all three. This is driven by some electrostatic attraction between the OH dipole of one hydroxyl group for the dipole of the second hydroxyl group. When do we want to call this attraction a hydrogen bond? What do we gain by not doing so for all three? If we understand that there is an energy continuum of hydrogen bonding, from weak to weaker, doesn’t that provide enough of a model to interpret and predict chemical structure and behavior?
References
(1) Lane, J. R.; Contreras-García, J.; Piquemal, J.-P.; Miller, B. J.; Kjaergaard, H. G. J. Chem. Theor. Comput. 2013, 9, 3263-3266, DOI: DOI: 10.1021/ct400420r.
InChIs
1: InChI=1S/C2H6O2/c3-1-2-4/h3-4H,1-2H2
InChIKey=>LYCAIKOWRPUZTN-UHFFFAOYSA-N
InChIKey=>LYCAIKOWRPUZTN-UHFFFAOYSA-N
2: InChI=1S/C3H8O2/c4-2-1-3-5/h4-5H,1-3H2
InChIKey=YPFDHNVEDLHUCE-UHFFFAOYSA-N
InChIKey=YPFDHNVEDLHUCE-UHFFFAOYSA-N
3: InChI=1S/C4H10O2/c5-3-1-2-4-6/h5-6H,1-4H2
InChIKey=WERYXYBDKMZEQL-UHFFFAOYSA-N
InChIKey=WERYXYBDKMZEQL-UHFFFAOYSA-N
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.