Fogueri, U. R.; Kozuch, S.; Karton, A.; Martin, J. M. L. J. Phys. Chem. A 2013, 117, 2269
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission
1

This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission
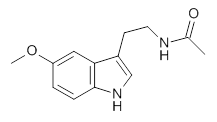
Conformational analysis is one of the tasks that computation chemistry is typically quite adept at and computational chemistry is frequently employed for this purpose. Thus, benchmarking methods for their ability to predict accurate conformation energies is quite important. Martin has done this for alkanes1(see this post), and now he has looked at a molecule that contains weak intermolecular hydrogen bonds. He examined 52 conformations of melatonin 1.2 The structures of the two lowest energy conformations are shown in Figure 1.
1
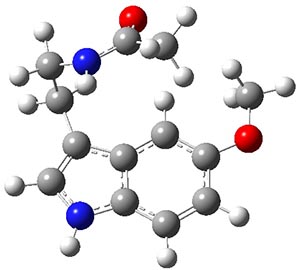
1a
| 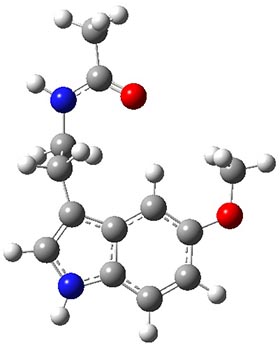
1b
|
Figure 1. Structures of the two lowest energy conformers of 1 at SCS-MP2/cc-pVTZ.
The benchmark (i.e. accurate) relative energies of these conformers were obtained at MP2-F12/cc-pVTZ-F12 with a correction for the role of triples: (ECCSD(T)/cc-pVTZ)-E(MP2/cc-pVTZ)). The energies of the conformers were computed with a broad variety of basis sets and quantum methodologies. The root mean square deviation from the benchmark energies is used as a measure of the utility of these alternate methodologies. Of particular note is that HF predicts the wrong ordering of the two lowest energy isomers, as do some DFT methods that use small basis sets and do not incorporate dispersion.
In fact, other than the M06 family or double hybrid functionals, all of the functionals examined here (PBE. BLYP, PBE0, B3LYP, TPSS0 and TPSS) have RMSD values greater than 1 kcal mol-1. However, inclusion of a dispersion correction, Grimme’s D2 or D3 variety or the Vydrov-van Voorhis (VV10) non-local correction (see this post for a review of dispersion corrections), reduces the error substantially. Among the best performing functionals are B2GP-PLYP-D3, TPSS0-D3, DSD-BLYP and M06-2x. They also find the MP2.5 method to be a practical ab initio alternative. One decidedly unfortunate result is that large basis sets are needed; DZ basis sets are simply unacceptable, and truly accurate performance requires a QZ basis set.
References
(1) Gruzman, D.; Karton, A.; Martin, J. M. L. "Performance of Ab Initio and Density Functional Methods for Conformational Equilibria of CnH2n+2 Alkane Isomers (n = 4-8)," J. Phys. Chem. A 2009, 113, 11974–11983, DOI: 10.1021/jp903640h.
(2) Fogueri, U. R.; Kozuch, S.; Karton, A.; Martin, J. M. L. "The Melatonin Conformer Space: Benchmark and Assessment of Wave Function and DFT Methods for a Paradigmatic Biological and
Pharmacological Molecule," J. Phys. Chem. A 2013, 117, 2269-2277, DOI: 10.1021/jp312644t.
Pharmacological Molecule," J. Phys. Chem. A 2013, 117, 2269-2277, DOI: 10.1021/jp312644t.
InChIs
1: InChI=1S/C13H16N2O2/c1-9(16)14-6-5-10-8-15-13-4-3-11(17-2)7-12(10)13/h3-4,7-8,15H,5-6H2,1-2H3,(H,14,16)
InChIKey=DRLFMBDRBRZALE-UHFFFAOYSA-N
InChIKey=DRLFMBDRBRZALE-UHFFFAOYSA-N
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
No comments:
Post a Comment