Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission
A quick note here on the use of computed NMR to determine stereochemical
structure. The Garg group synthesized two “oxidized welwitindolines”,
compounds 1 and 2.1 The relative stereochemistry at the C3 position (the carbon with the hydroxy group) was unknown.
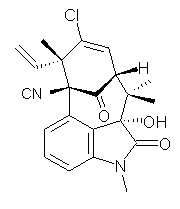
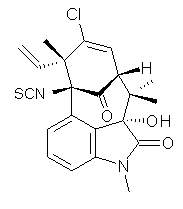
1 2
Low energy gas-phase conformers of both epimers of 1 and 2 were
optimized at B3LYP/6-31+G(d,p). (These computations were done by the
Tantillo group.) See Figure 1 for the optimized lowest energy
conformers. Using these geometries the NMR chemical shifts were computed
at mPW1PW91/6-311+G(d,p) with implicit solvent (chloroform). The
chemical shifts were Boltzmann-weighted and scaled according to the
prescription (see this post) of Jain, Bally and Rablen.2 The computed chemical shifts were then compared against the experimental NMR spectra. For both 3 and 4, the 13C NMR shifts could not readily distinguish the two epimers. However, the computed 1H chemical shifts for the S epimer of each compound was significantly in better agreement with the experimental values; the mean average deviation for the S epimer of 2 is 0.08 ppm but 0.36ppm for the R epimer. As a check of these results, DP4 analysis3 (see this post) of 2 indicated a 100% probability for the S epimer using only the proton chemical shifts or with the combination of proton and carbon data.
References
(1) Quasdorf, K. W.; Huters, A. D.; Lodewyk, M. W.; Tantillo, D. J.;
Garg, N. K., "Total Synthesis of Oxidized Welwitindolinones and (-)-N-Methylwelwitindolinone C Isonitrile," J. Am. Chem. Soc. 2011, 134, 1396-1399, DOI: 10.1021/ja210837b
(2) Jain, R.; Bally, T.; Rablen, P. R., "Calculating Accurate Proton Chemical Shifts of Organic Molecules with Density Functional Methods and Modest Basis Sets," J. Org. Chem. 2009, 74, 4017-4023, DOI: 10.1021/jo900482q.
(3) Smith, S. G.; Goodman, J. M., "Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability," J. Am. Chem. Soc. 2010, 132, 12946-12959, DOI: 10.1021/ja105035r
(2) Jain, R.; Bally, T.; Rablen, P. R., "Calculating Accurate Proton Chemical Shifts of Organic Molecules with Density Functional Methods and Modest Basis Sets," J. Org. Chem. 2009, 74, 4017-4023, DOI: 10.1021/jo900482q.
(3) Smith, S. G.; Goodman, J. M., "Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability," J. Am. Chem. Soc. 2010, 132, 12946-12959, DOI: 10.1021/ja105035r

This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
No comments:
Post a Comment